License: Boltz-2 is open source and free for academic and commercial use under an MIT license. Please refer to the license for full terms.
This constraint is open source. Any third-party models, product names, or trademarks referenced are the property of their respective owners, and Proto is not affiliated with them.


[0.0, 1.0]
against configurable targets/tolerances and combined via weighted averaging;
default weights are chosen by complex type.
API Reference
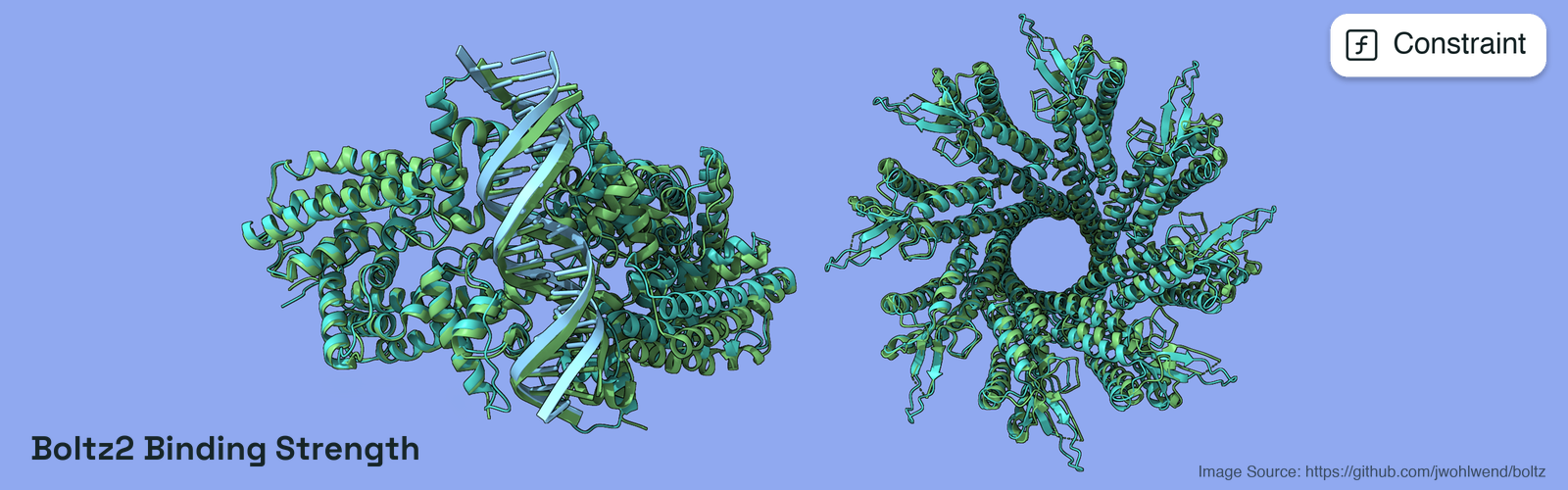
Configuration for Boltz binding strength constraint.This class defines configuration parameters for evaluating protein-protein,
protein-ligand, and protein-nucleic-acid (DNA/RNA) binding using Boltz, a
biomolecular structure prediction model. A common use case is scoring a
protein occupying a nucleic-acid target (e.g. a repressor bound to a DNA
operator) via interface confidence. Boltz predicts complex structures and
provides confidence metrics for binding quality, interface accuracy, and
overall structure reliability. The constraint evaluates these metrics against
target values to assess binding strength and quality.The constraint uses a penalty-based scoring system where each metric is evaluated
against its target value and tolerance. Metrics are classified as “higher is better”
(e.g., interface confidence scores) or “lower is better” (e.g., predicted distance
errors). Penalties are combined using weighted averages, with default weights
chosen by complex type (monomer, protein-ligand, or multi-chain — the latter
covering both protein-protein and protein-nucleic-acid complexes).
Metric interpretation:
- iptm/ligand_iptm/protein_iptm: Interface confidence (0-1). Higher = better binding prediction. Values >0.8 indicate confident binding interfaces.
- complex_iplddt: Interface per-residue confidence (0-1). Higher = more reliable interface residue predictions.
- complex_plddt: Overall structure confidence (0-1). Similar to ESMFold pLDDT.
- ptm: Overall structural accuracy (0-1). Similar to ESMFold pTM.
- complex_ipde/complex_pde: Predicted distance errors in Ångströms. Lower = more accurate structure. Values <3 Å indicate high accuracy.
- confidence_score: Boltz’s aggregate confidence combining multiple factors.
object
Target values for ‘higher is better’ metrics.
object
default:"{'complex_ipde': 2.0, 'complex_pde': 2.0}"
Target values for ‘lower is better’ metrics.
object
Tolerances for higher-is-better metrics; once a value falls this far below target, penalty hits 1.0.
object
default:"{'complex_ipde': 2.0, 'complex_pde': 3.0}"
Tolerances for lower-is-better metrics (Å); once exceeding target by this much, penalty hits 1.0.
object
Weights for combining penalties
boolean
default:"True"
Whether to include confidence_score in penalty calculation (adds weight 0.10)
enum
default:"total_penalty"
Component to return: ‘total_penalty’ (weighted combination) or specific metric nameOptions:
total_penalty, iptm, ligand_iptm, protein_iptm, complex_iplddt, complex_plddt, complex_pde, complex_ipde, confidence_score, ptmBoltz2Config
Boltz2 configuration for structure prediction.
ReturnsConstraintOutput
Per-complex score in [0.0, 1.0] (0 = perfect
binding). Predicted Boltz structure is attached to the first slot of
each complex. metadata carries boltz2_binding (a list of
dictionaries, one per evaluation):penalty: Float overall constraint score (0.0-1.0)metrics: Dictionary of all raw Boltz metrics (iptm, iplddt, etc.)penalties: Dictionary of individual metric penalties before weighting
Usage
python